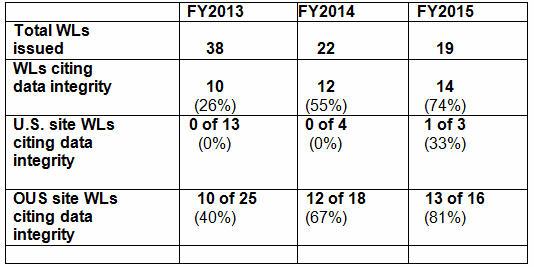
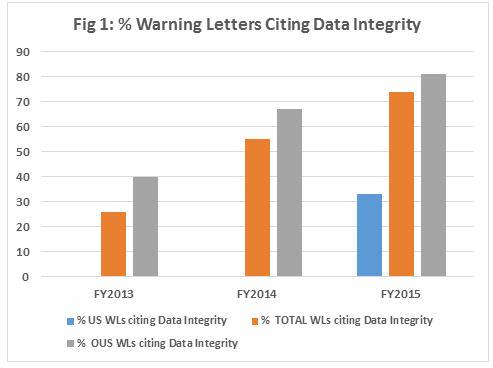
Along with providing monitoring and validation systems, we often delve into issues that arise for our customers when they are interpreting regulations and guidance. We receive many questions on 21 CFR Part 11 and Annex 11. In this article we we offer some background and a brief overview of three focal points of both of the "Elevens" including: System Controls, Validation and Archiving.
It's important to note that Part 11 is a requirement in the US, whereas Annex 11, which applies to the EU, is a guidance document only. By the way, we are assured by those in the know that the "11" in the titles are incidental. Someone please correct us if we've been misinformed!
Background:
Computerised systems — crucial to pharmaceutical, medical device, and biotechnology manufacturing and distribution operations — differ from paper-based systems and manual systems traditionally used for creating and archiving records are becoming rare.
This is (partly) why the FDA and EMA created 21 CFR Part 11 and Annex 11. But the real basis for of "the elevens" is to ensure that the quality and safety of drugs and biologicals do not suffer as a result of computerized systems replacing a manual system.
Annex 11 states:
"Where a computerized system replaces a manual operation, there should be no resultant decrease in product quality, process control or quality assurance. There should be no increase in the overall risk of the process."
The FDA similarly says that the purpose of Part 11 is to make sure electronic records are:
"...trustworthy, reliable, and generally equivalent to paper records."
The FDA's statement entails the fact that for generations, paper records were all we had to depend upon to ensure that processes and conditions that preserved the safety and quality of drugs were performed properly.
Both Part 11 and Annex 11 remind us of the importance of safety, and address the need to set up standards to make ink and electronic or digital signatures equivalent in their effect.
Controls: Human Readable, Unmodifiable, Authorized
Both Part 11 and Annex 11 include the following elements:
• Validation
• Human Readable Copies
• Protection and retention of records
• Audit trails
• Restricted access for authorized users only
• Authority checks
• Device checks
• Training
• Written procedures
• System documentation
One glimpse at the list shows that each is in some part a method for controlling the function and outputs of a system.
In 21 CFR Part 11 "Controls for Closed Systems" states:
(b) "The ability to generate accurate and complete copies of records in both human readable and electronic form suitable for inspection, review and copying by the agency."
The fact that there is a specific regulation regarding human-readable copies demonstrates how far technology has evolved. It would be hard today to imagine a user-friendly system that did not allow for the printing of documents and data.
For most monitoring systems, the records of interest are the actual historical monitoring values. And creating a human-readable copy likely means that historical data and event logs may be printed out in a secure format.
In 21 CFR Part 11 (e) in "Section 11.10 - Controls for Closed Systems we read:
"Use of secure, computer-generated, time-stamped audit trails to independently record the date and time of operator entries and actions that create, modify, or delete electronic records. Record changes shall not obscure previously recorded information. Such audit trail documentation shall be retained for a period at least as long as that required for the subject electronic records and shall be available for agency review and copying."
In terms of your environmental monitoring applications, this simply means that, in order for your records to comply, the electronic records (data and events) cannot be manually modifiable or deletable.
In addition, the Audit Trail of your system needs to capture any changes to metadata (schedules and report templates) and configuration data without obscuring earlier entries. If your system doesn’t allow any changes to values once recorded, it complies with those sections of both Part 11 and Annex 11.
As a closed system, your monitoring system needs to limit access to only "authorized individuals." That's item (d) in Section 11.10 of Part 11. Typically this means that all who have access to the system have a distinct username and secure password. Often a system will integrate with your OS authentication to leverage commonly used, pre-existing password management tools.
Validation: Prove it Works & Document your proof
With regard to validation, Part 11 lays out the need for validation in the first item of Section 11.10: "Controls for Closed Systems":
(a) "Validation of systems to ensure accuracy, reliability, consistent intended performance, and the ability to discern invalid or altered records."
In Annex 11, the "Project Phase" section covers validation in eight fairly brief, fairly straightforward points:
Use risk assessment to justify standards, protocols, acceptance criteria, procedures and records
Use correct change control documentation practice to report deviations
Keep an Inventory of your computerised systems with descriptions, interfaces, processes, software and hardware
Use risk assessment to assess the GMP impact of User Requirements Specifications
Audit suppliers’ Quality Management Systems
Validation of computerised systems covers the entire life-cycle of the system
Document testing
Validate data transfers
We see that Annex 11 is more explicit in recommendations for validation.
Archiving: Validated, Secure, Accessible
The protection and retention of records, as described in Part 11's Section 11.10 (c) means that whatever data your system produces, it may not be altered. In addition, to meet the requirement of "ready retrieval throughout the records retention period," your data needs to be archived in a way that it can be conveniently accessed – you don’t want to make an auditor wait,
If you store the data permanently within your database, the database should be designed in such a way system's performance should not degrade as the database grows over time. How you meet this requirement of 21 CFR Part 11 may have as much to do with the design of your system as it does with your system administration policies around archival.
In Annex 11 under “Data Storage” it is recommended that data be securely stored, and backed up BOTH physically and electronically and regularly checked for accessibility, readability and accuracy. Annex 11 also mentions is the need to validation data restoration abilities of the system.
Part 11 and Annex 11 were introduced to address the key differences between computerized and manual systems and make electronic records equivalent to paper records as evidence of quality process execution. Today, most environmental monitoring systems used in GxP compliant firms are inherently aligned with the requirements of both the "Elevens." However, the risk comes not from the systems themselves, but in how they are implemented and maintained.
Although your monitoring system probably included User and Administrator manuals and CDs that are fairly easy to integrate into your Document Management System, the procedures that control documentation are still your responsibility. As good as your monitoring system may be (and, if you have a Vaisala system, it's awesome!), only the procedures of your Quality System keep you in compliance with Part 11/Annex 11.
We didn't cover Electronic Signatures because that's a specialized software function. Specialty enterprise software exists for implementing Electronic Signatures within an Electronic Document Management System (EDMS). For Vaisala's monitoring solution, we use a "hybrid" system, which means it uses electronic records combined with handwritten signatures, wherein the PDF outputs can be imported into and EDMS.